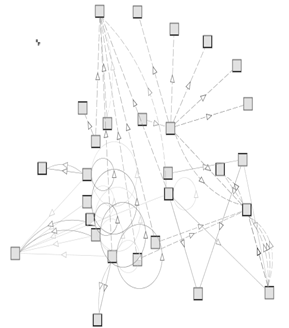
Figure 1: Selection of the first 3 levels in the GlycO hierarchy
|
Copyright is held by the World Wide Web Conference Committee (IW3C2). Distribution of these papers is limited to classroom use, and personal use by others. WWW 2006, May 23-26, 2006, Edinburgh, Scotland. ACM 1-59593-323-9/06/0005. |
As part of the Integrated Technology Resource for biomedical glycomics, established by National Center for Research Resources, a team of biologists and biochemists at the Complex Carbohydrate Research Center (CCRC), University of Georgia (UGA) are working towards the standardization of experimental protocols for high-throughput glycoproteomics research. To enable and support this endeavor, bioinformatics researchers from the Large Scale Distributed Information Systems (LSDIS) lab, UGA and CCRC are working on building a semantic framework to solve the attendant informatics issues. Two ontologies, GlycO and ProPreO (both available publicly, see: http://lsdis.cs.uga.edu/library/resources/) form the foundation of this framework. GlycO is a glycoproteomics domain ontology for modeling the structure and functions of glycans, enzymes and pathways. ProPreO is a process ontology for modeling the complete glycoproteomics experimental lifecycle to enable ontology-mediated data classification, storage, retrieval and provenance.
Ontologies are being increasingly used by the biological community as standard knowledge representation models for integrating, sharing and managing data and information. Nonetheless, many of the available and used biological ontologies are not logically rigorous. It is important to note that, in addition to storing and sharing of biological data, computational reasoning over data, using ontology as the reference, is expanding rapidly [32]. Hence, inherent inconsistencies, contradictions or incorrectness in the modeling of the biological domain can be detrimental to computational applications. Many biological ontologies have incorrectly determined classes, incorrect or inappropriate naming schemes, and have ill defined relationships between concepts. Such deficiencies in the specific case of MGED, a highly visible ontology, are discussed in detail in [32].
We adopted OWL-DL [8] for ontology development, carefully balancing the pros and cons of expressiveness and computability. Most important for us were value restrictions and exact cardinality restrictions that we can express in OWL-DL, but not in OWL-Lite. However, in some cases we experienced the limitations of OWL-DL. Especially the strict distinction between schema data and instance data created problems. The next member of the OWL family, OWL-Full, is a syntactic and semantic extension of RDFS. It is less restrictive than the other flavors, but not decidable. For an Ontology that is used for reasoning tasks, consistency is mandatory. Using a language that is not decidable would not permit automatic consistency checking. The reasoner could produce wrong results even for simple queries.
Another important aspect in a biological ontology is the role of relationships. A simple taxonomy of concepts is inadequate to model the richness and extensiveness of the relationships between biological entities, chemical entities and the experimental processes aimed at revealing them. The absence of these relationships handicaps the extensibility and most critically the usability of an ontology. The expressiveness of the GlycO and ProPreO ontologies is due in part to the incorporation of a large number of instances and explicit specification of relationships between the instances. The instances link real-world entities to the schema and are essential for the functionality of an ontology-driven application. The process of populating such highly connected ontologies is a considerable challenge, and it was necessary to develop new methods to populate the GlycO and ProPreO ontologies.
1.1. Contributions and Outline
§ In this paper, we focus on original approaches used in developing the schemas of the GlycO and ProPreO ontologies. In the case of GlycO, there are thousands of glycans, formed of many constituent sub-entities, so-called residues, which need to be captured. This is accomplished by using a canonical representation model, based on the GlycoTree [37]. In ProPreO, in addition to modeling an end-to-end glycoproteomics experiment, a semantic data provenance scheme is being implemented using a set of Universal Resource Identifiers (URI). This composite URI is built using modular blocks of URIs, which are concepts in ProPreO. A particular URI block may be accessed in a single-step, and interpreted using ProPreO as the reference. Hence, this forms a flexible semantic data provenance scheme. As part of the schema design for the two ontologies, we also focus on the importance of modeling relationships between concepts.
§ We also discuss the approaches used in populating these large ontologies with real-world information in the form of instances from multiple public databases including KEGG [9] [19], SweetDB [12] [20] and intra-lab data collections such as lists of human tryptic peptides generated at the CCRC [11] [5]. We describe the use of GLYDE (GLYcan Data Exchange) [27] [28], an XML-based glycan data representation standard for populating GlycO. In case of ProPreO, we discuss our approach to populating a complex ontology with extremely large datasets using a dual-level instance base in which the experimental data (some having a size of 500MB), are stored in a separate location. These large data sets are logically integrated into the instance base when necessary for use by a reasoning tool.
§ Finally, in addition to discussing the use of structural metrics [38] to compare the two ontologies with the MGED ontology and some of the ontologies listed at OBO; we also focus on the application of these ontologies as part of the semantic informatics structure for glycoproteomics research.
It has been recognized that there are no widely accepted guidelines for developing domain (hence also biological) ontologies [33]. Experiences presented in this paper provide insights into the challenges in developing such a framework.
The rest of the paper is organized as follows: section 2 presents the development of the two ontology schemas and the population of the ontologies. Section 3 discusses the evaluation of GlycO and ProPreO using multiple structural metrics. Section 4 discusses the application of these ontologies as part of the NCRR glycomics bioinformatics project [21]. Section 5 and Section 6 discuss related work and conclusion respectively.
2. ONTOLOGY DEVELOPMENT
In this section we detail the schema development and population aspects of GlycO and ProPreO. We used the Protégé ontology editor [23] for the schema design. Envisioning ontologies with a very large number of instances, we used a different route for populating them. Semagix Freedom, which is a commercialization of research in the LSDIS lab [30], was used to extract potential instances from databases and the World Wide Web. Additional software developed at the LSDIS lab was used to transform the extracted textual information into more expressive OWL-descriptions.
Different ontologies focus on different domains, even different views of the same domain. Ontologies are also developed in light of different applications and consequently with the logical rigor that is appropriate for these applications. For example, the CYC [25] ontology is developed with extreme logical rigor, in order to give intelligent agents comprehensive world-knowledge. The TAP [7] ontology, SWETO [1] or the Gene Ontology GO [2] on the other hand, have a relatively simple logical model. Their applications include disambiguation, annotation and knowledge discovery. Since both GlycO and ProPreO make extensive use of OWL-DL, their expressiveness lies between CYC and ontologies based on "lighter" models. An interesting reference point is the strategy used for population and its attendant costs. For CYC, each concept is manually generated, which makes the development very expensive. The same holds, so far, for GO. Since TAP is populated by crawling and scraping websites, and SWETO is populated by commercial knowledge extraction and disambiguation technologies that are part of Semagix Freedom, the population related costs are significantly lower. However, the TAP and SWETO schemas are not very expressive. In ProPreO and GlycO we adopted a different strategy. A very expressive schema was generated, but the crucial part of populating the schemas involved three different approaches, which required the development of additional tools as described later.
2.1 Ontology structure
The following sections discuss the structural aspects of the two ontologies, focusing on their level of granularity. The high degree of specialization (fine granularity) is a key quality of these ontologies that make them useful in the target domain.
2.1.1 GlycO
The aim of glycoproteomics is to understand the interaction of glycans with genes and proteins, and the cellular processes in which they participate. Since no unified, formalized description of this complex domain had previously existed, the GlycO ontology had to be built from scratch. Our goal was to design a schema that is expressive enough to semantically model the subtle differences in glycan structure that modulate their biological functions. In this context, GlycO is meant to be more than a controlled vocabulary; its intention is to be used for reasoning in scientific analysis and discovery.
Initially, we analyzed the glycoproteomics domain broadly, collected terms, and examined the way these terms are used by scientists. It turns out that the informal usage of the 'is_a' relationship, as in "a glycan is a carbohydrate", implies a hierarchy of concepts with multiple inheritances. We wanted to keep the "colloquial" use of the glycoproteomics terminology consistent with the ontology, while also adding more accurate descriptions. In addition, the is_a relationship between classes assures a very intuitive way of doing subsumption-based reasoning. There are many ways of classifying monosaccharide residues, which are the building blocks of glycans. For example, it is possible (and equally valid) to classify them according to the number of carbon atoms in the monosaccharide or as a structural variant. That is, a b-d-Glcp residue can be identified as both a hexosyl residue (with 6 carbons) and an aldosyl residue (embodying the aldo-structural variant). Other classifications are possible and the commonly used terminology suggests that a single monosaccharide residue can embody more than one structural variation, (e.g., keto and deoxy), along with a ring form (e.g. pyranosyl), an overall configuration (e.g. gluco), an anomeric configuration (e.g. b) or an absolute configuration (e.g., d). We account for all of these properties by allowing a particular monosaccharide residue to inherit from several super classes. Whether this directed acyclic graph is explicitly designed or inferred is secondary. For example, the absolute configuration d and subsumption by the superclass residue are necessary and sufficient properties of the class d-residue. A reasoner will automatically subsume any residue class that has the absolute configuration d under the class d-residue. A hierarchy with multiple inheritance will almost always automatically arise when a more sophisticated logical description of classes is used alongside restricting conditions.
Our first level of abstraction contains the three classes Chemical Entity, Chemical Property and Reaction. This is an appropriate starting point in that we can subsume these classes under the SUMO [36] concepts Object, Attribute and Process. From there, we define a finely grained class hierarchy (see Figure 1 for a selection of the first 3 levels of the GlycO hierarchy).
|
Figure 1: Selection of the first 3 levels in the GlycO hierarchy |
With 11 levels, GlycO has a deeper hierarchy than many other domain ontologies. When designing a hierarchy of concepts that reflect the natural occurrence of objects, we are restricted to what actually exists. We therefore relaxed Schulze-Kremer's [29] requirement that each subclass should be distinguished from its super class by exactly one discriminating criterion.
The classification scheme in GlycO is designed to extend this idea of rigorous restrictions to all of the monosaccharide residues within the glycan. For the current version, which focuses on N-glycans, this is accomplished by defining a tree structure of canonical residue entities that subsumes most N-glycans. That is, almost all of the known N-glycan structures can be completely specified by choosing a subset of the nodes of this tree. This subset forms a connected subtree that includes the root residue. This tree (known as GlycoTree) has been previously described [37], and we have formalized that structure as a collection of interconnected, canonical residue instances in GlycO.
The hierarchy of concepts is one aspect of semantics captured in an ontology, but the addition of other relationships is required to realize a useful and powerful model. Relationships have been seen as the key to semantics for some time (see review in [31]). A concept by itself might be useful for a human observer, but only if he can look at it within a context of other concepts. The human infers related concepts according to his background knowledge. For a machine this background knowledge needs to be stated explicitly. The authors of [32] raised the issue that MGED contained too many named relationships that impede the computational use of the ontology. We agree insofar as those relationships should be unambiguous; no two different named relationships should have the same semantics. Also, the ontology becomes less general and it becomes harder to map the relationships to other ontologies for the purpose of merging or interaction.
We address dilemma of generality versus computational complexity by making use of a relationship hierarchy, modeling the relationships from more general down to more specific. Upper level relationships are e.g. has_part or affects and their inverses. Inheriting lower level relationships restrict domains and ranges of the upper level relationships. For example, has_carbohydrate_residue is essentially a has_part relationship, but its domain is restricted to glycan and its range is restricted to carbohydrate_residue. A reasoner will be able to map this relationship to a more general relationship in a different ontology.
2.1.2 ProPreO
We developed the ProPreO ontology as a formal representation of proteomics processes and attendant data. The two critical aspects of any '-omics' experiment are the identification of biological entities and their quantification i.e. 'what is it?' and 'How much of it is there?' It is extremely difficult, especially in a high-throughput environment, to answer these questions by querying datasets that are heterogeneous, developed by multiple researchers who use different methodologies, parameters, and data formats. Although provenance can form a foundation to compare different datasets and enable researchers to repeat experiments and track the attendant data [40] [41], syntactic data provenance is inadequate to support such queries.
To solve these challenges we are developing a semantic Universal Resource Identifier (SemURI) scheme as an integral part of ProPreO. By semantic URI we mean an URI that lexically incorporates semantic description by succinctly representing an ordered list of concepts that are part of ProPreO. This framework will facilitate ontology-mediated data provenance, dataset annotation using concepts from the schema, and the generation of separately stored metadata, which may be used by computational tools to compare and correlate datasets in the relevant context. In case of experimental data, the context for comparison and correlation is provided by multiple factors such as the origin of the sample (e.g., malignant or benign tumor cells), experimental methods used in the generation of the data (e.g., the chromatography method used to separate peptides), the settings of individual instruments (e.g., the laser intensity of an ion source), or the database used in identification of peptides. The starting point in the development of ProPreO was the Pedro UML schema [39], which models four stages of experimental proteomics, namely Sample Generation, Sample Processing, Mass Spectrometry and MS Results Analysis. However, the goals of ProPreO are distinct from those of Pedro UML schema [39], and hence these four stages are not defined as top-level concepts in ProPreO. We iteratively evolved the current top level concepts of ProPreO through multiple use cases of applications listed above.
In the following paragraphs, we discuss the top level concepts of ProPreO and illustrate its ability to describe experimental hardware, data processing applications, laboratory tasks, computational tasks, parameter sets, and the resulting data. We also discuss its extensibility, which allows new classes of the above listed concepts to be included in the ontology. This reflects the state of real biological experimental protocols, i.e. they must be sufficiently dynamic to keep pace with new technologies and paradigms.
data - We have modeled the various types of datasets generated at different phases of a glycoproteomics experiment. For example, data generated by analytical techniques, such as mass spectrometry exist in multiple forms. Typically, the 'raw data' initially generated by a mass spectrometry instrument is in a proprietary format. Subsequently, it is processed to generate another subclass of data, i.e., a list of glycopeptides. The metadata required to provide the relevant experimental context for comparison of processed datasets includes parameter values for the tasks that generated them. The rich set of relationships in ProPreO (see Figure 2) allows data instances to be compared by associating them with instances of other relevant concepts such as 'parameter_lists'.
data_processing_tool - There are many standard and locally developed software applications used to generate or process data at various stages of the experiment. Metadata semantically annotates data instances, associating each with specific software applications and forming an appropriate context for interpretation and processing.
hardware - The 'hardware' concept includes the two subconcepts 'instrument' and 'instrument_component'. This enables ProPreO to capture metadata describing the states of the various components of the instrument that generated a given instance of data. This metadata is also necessary to determine whether two datasets can be directly compared.
For example, the instrument component HPLC_diode_array_detector has two properties that specify the range of wavelengths that are accessible to the detector ('has_wavelength_detection_max' and 'has_wavelength_detection_min'). These properties allow a reasoner to infer that data generated using an ultra-violet (UV) detector cannot be directly compared with data generated using a visible light detector.
molecule - This defines the very broad classes of molecules that are analyzed in a proteomics experiment; for example, glycans, proteins and peptides. These classes are themselves defined (at least in part) by their equivalence to analogous classes in more specialized ontologies such as GlycO. ProPreO extends these class definitions by adding properties that provide an experimental context of reference. This framework allows, e.g., a 'peptide' to be associated with its 'experimental_chemical_mass', a property that may not be defined in the referenced ontology.
organism - This class describes the taxonomic classification of a biological species, again by reference to a more specialized ontology, providing the biological context of a given sample instance.
|
Figure 2: Relationships in ProPreO excluding regular relationships 'is-a' and 'has-instance' (generated using Jambalaya plug-in with Protégé) |
parameter_list- Each instance of data has a set of parameter values that are associated with its generation. These include the instrumental settings, environmental parameters and variable setup parameters used by software applications to process data. ProPreO models parameters relating to database searching, HPLC runs and mass spectral analysis.
The experimental context of a parameter (which is a type of data) can be inferred by its inclusion in a particular parameter_list. Furthermore, parameters are subclassified according to their relationships to specific experimental task, providing a rich framework for analyzing their relevance with regard to experimentally obtained data.
task - A glycoproteomics experiment can be viewed as a set of human-mediated or automated tasks that generate real-world samples or data to be used as input for the next step. Classification of tasks is a key feature underlying our implementation of the SemURI scheme (described in previous sections) for semantic data provenance.
2.2 Ontology population
As already stated, the population of an ontology with instances from the real world, representing the concepts defined at the schema level forms a critical aspect of the knowledge captured by an ontology. In the following sections, we describe the various challenges faced when populating GlycO and ProPreO, and the approaches employed to overcome them.
2.2.1 GlycO
Once a sufficient description of the domain was given by the developed schema, we started populating the ontology with instances. The population is done both manually and automatically. A small number - 158 - of rigorously described concepts, such as monosaccharides, which function as building blocks of more complex carbohydrates, have been inserted manually by the domain expert to assure accuracy and comprehensive description at this important level. The number of monosaccharides is very limited; hence the manual population of this part of the ontology is also the most efficient way.
Numbers of instances of other biological and biochemical structures that can populate GlycO are not as modest. Thousands of Glycans, Proteins and Genes make their virtual appearance in many different databases. In order to harvest this data, we used the Semagix Freedom toolkit that allows extraction of data from semi-structured web pages and database driven web sites. Simply collecting this information is not enough, since database schemas are usually shallow and categorization is done by keywords rather than by a class hierarchy. Hence instances have to be classified after extraction from the source. If the class hierarchy is, amongst other restrictions, value restricted, keywords can be used to aid the classification of the instance data. Since, in the case of GlycO, the classification is finer than the keyword-based classification in most databases, the instances have to be classified according to their structure. The conversion of the glycan structure into the LINUCS [3] [10] compatible GLYDE [27] format provides the initial step. The instance information is then analyzed according to GlycoTree [37]. The glycan is split into its residues and each residue is categorized as a contextual residue, which provides a canonical residue which is part of the GlycoTree.
In order to have source data of highest quality, we chose to extract instances from different databases and compare them during the encoding phase. The databases used were KEGG [9] [19], SweetDB [12] [20] and CarbBank [11] [5], which was developed at the CCRC.
Populating an ontology automatically from several sources is both an opportunity and a challenge. In order to get the highest quality and quantity of knowledge, potentially more than one source has to be often consulted for every instance put in the ontology. Each source might focus on different criteria (or provide different or overlapping properties associated with a concept) and leave out others that we still want to insert in our knowledge base. Hence the knowledge extractor has to differentiate between new instances and those that have been inserted before and can be enriched with new information from a different database. For this, the extractor needs to have sophisticated entity disambiguation techniques. Most databases use unique proprietary accession numbers for their entries, so a disambiguation across databases by key is not always possible. Different naming conventions prohibit disambiguation by name. Many different glycan structures have the same elemental composition (meaning the same number of each of its atoms and hence also the same molecular mass). Finally the IUPAC [17] notation for glycan structure in its simple form is not unique, so it cannot be a discrimination criterion either.
The easiest way to disambiguate in our domain was to find a common link to a CarbBank accession ID for the particular glycan. CarbBank is still one of the most comprehensive and most referenced collections of glycan structures and related publications.
However, since the curation of CarbBank was discontinued, not every glycan has a representation in CarbBank. For these new cases, the IUPAC structure of the glycan is sent to the SweetDB web based application [12] to convert it into the unique representation of the LINUCS format. This unique identifier allows a reliable disambiguation in the absence of other discriminating data. Since also the IUPAC to LINUCS conversion is purely syntactical, ambiguities in the naming of the residues can lead to ambiguities here. Using the LINUCS to GLYDE conversion Web Service [27], an unambiguous XML description of the glycan is built, which is then converted into the GlycoTree [37] based representation in the ontology. This is an example of a domain specific disambiguation approach where general techniques of disambiguation would most likely fail (see Figure 3).
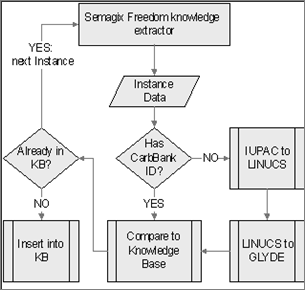 Figure 3: Ontology population workflow for
GlycO
Figure 3: Ontology population workflow for
GlycO
Another major obstacle that has to be overcome when a highly expressive schema is defined is that of incomplete knowledge. Some properties of a class might be set as required in the schema, because real-world entities that belong to this class would have these specific properties. However, these properties might not be explicitly stated in the knowledge source, but implicitly available in the glycan structure or otherwise deducible from known facts. Since rule base inference is not a part of the OWL framework, this deduction is best being done prior to adding the instance to the ontology with special tools, or, where appropriate, using SWRL [13] rules on top of OWL. The structural representation of glycans is best done with the specialized tools we described earlier. Other relationships, such as is_precursor_of can be added using SWRL rules. E.g. if enzyme E is involved in a reaction that forms glycan Y by adding a carbohydrate residue to glycan X, then X is the precursor of Y.
2.2.2 ProPreO
The experimental datasets generated by high-throughput glycoproteomics experiment are extremely large. For example, one mass spectrometry sample run generates 500 MB of data and in a typical research lab; hundreds of such samples are run in the course of one project. Hence, to physically store all instances of these experimental data in the knowledge base of ProPreO was an impractical choice. But, it was equally important to enable a data management application or reasoning tool to have access to all relevant datasets within the framework of the ontology schema. This involved a mechanism by which we could present a logically unified view of the instance base, without physically storing the large volumes of data in the ontology. Using the strategy of dynamically loaded libraries in programming languages that are resolved at runtime by the compiler, we created a scheme of using Universal Resource Identifiers (URI) as 'pseudo-instances' in the knowledge base of an ontology. These URIs are pointers to the physical location of experimental datasets. Similar to programming languages, these URIs are resolved at runtime and the actual experimental data in integrated into the ontology. This may be performed by the tools or a wrapper application that expands the ontology with the experimental datasets in place of the URIs and presents it to the tools.
|
|
|
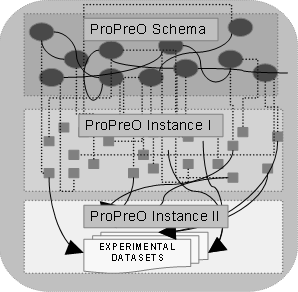
Figure 4: The figure illustrates the two levels of instance base of ProPreO. |
Proteomics experimental data also involves a set of instances that is referred to recurrently by other data sets and the population size of these common datasets is relatively small. For example, the set of human tryptic peptide sequences are instances of the concept 'peptides' that are generated from proteins by trypsin proteolytic enzyme. Therefore, it was intuitive to physically store these instances in the ontology knowledgebase which may be referred to by other experimental data sets.
Hence our solution to these two orthogonal requirements for the population of instances in the ProPreO knowledge base involved the use of two levels of instance base (see Figure 4):
Level 1: This is the regular instance base of an ontology consisting of extracted instances of the recurrent dataset, namely the human tryptic peptide sequences. An internal data collection at the CCRC had the relevant data. Hence, we used customized scripts to extract these structured data and populated the ontology.
Level 2: We are using a dedicated facility capable for storing multiple terabyte data, at the University of Georgia to store all data generated by this project. The URI used to locate the datasets is separate from the URI scheme we are using for the ontology-mediated data provenance. The URI generated by each datasets stored in the central repository is manually added to the ProPreO instance base.
Although at this time we are not using experimental data for reasoning purposes, as this semantic data management framework is further developed, we will need to access the experimental datasets within an ontological framework for information retrieval and ultimately knowledge discovery.
3 ONTOLOGY METRICS
Using OntoQA [38], we have compared GlycO and ProPreO with a set of ontologies listed at OBO [14] and the MGED ontology (see Table 1). The set of structural metrics used in this evaluation have been chosen to give a numerical account of some of the characteristics of the tested bio-ontologies. As mentioned in the introduction, it is not possible to have an algorithmic procedure for the development of a good ontology. It is possible, to a much lesser extent, to measure the "goodness" of an ontology with some numerical values. The purpose of these metrics is to give an account of the structural composition of two ontologies.
Table 1: Results of comparison with ontologies listed at OBO. The comparison is on the total number of concepts, average number of concepts subsumed by a concept and number of relationships per concept (connectivity).
|
Ontology |
No. of Terms |
Avg. sub- terms |
Connectivity |
|
GlycO |
382 |
2.5 |
1.7 |
|
ProPreO |
244 |
3.2 |
1.1 |
|
MGED |
228 |
5.1 |
0.33 |
|
Biological Imaging methods |
260 |
5.2 |
1.0 |
|
Protein-protein interaction |
195 |
4.6 |
1.1 |
|
Physico-chemical process |
550 |
2.7 |
1.3 |
|
BRENDA |
2,222 |
3.3 |
1.2 |
|
Human disease |
19,137 |
5.5 |
1.0 |
|
GO |
200,002 |
4.1 |
1.4 |
For example, we believe that relationships between concepts are of critical importance in an ontology for use in biological domain. Hence, we use these values to provide a quantitative aspect of GlycO and ProPreO based on a set of metrics. We do not claim that these set of metrics are comprehensive or exclusively form the basis for evaluation of ontologies.
We see that GlycO and ProPreO have an intermediate number of terms when compared to the rest of the OBO ontologies, which indicates that the information they contain is of an adequate size in the biological domain. The average number of sub terms per term in all ontologies is relatively similar, which also indicates that GlycO and ProPreO have an adequate information distribution across the different levels of the term inheritance tree. It can be also seen that GlycO terms have higher connectivity to other terms in the ontology when compared with the other ontologies. This indicates that the interactions between the terms in GlycO are higher than that of the other ontologies, while the number of interactions between ProPreO terms is relatively similar to the OBO ontologies.
Figure 5 shows the average fan-out (average number of subclasses per class) and the height of the inheritance trees of ProPreO, GlycO and MGED. GlycO, a highly specialized domain ontology, is deep and narrow, while MGED's purpose is to give a broader description of microarray gene experiments. It would be of empirical interest to see whether most ontologies follow this trend i.e. the more specialized the application area of the ontology is, the deeper and narrower it is designed. This is certainly not a universal rule, because it is easy to construct ontologies that would defy it. But it is possible that this is simply "how we design ontologies".
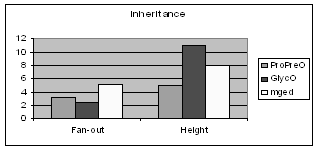
Figure 5: Comparison of GlycO and ProPreO with MGED on Inheritance specific metrics
The number of properties in an ontology indicates the richness of the relationships that can possibly combine the different types of objects in the ontology. ProPreO, which aims at very carefully describing experimental data and processes, allows for the strongest connectivity of its instances, as Figure 6 shows.
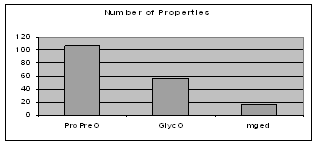
Figure 6: Relationship richness in GlycO, ProPreO and MGED ontologies
4 ONTOLOGY APPLICATIONS
In the following sections, we describe two specific applications of GlycO and ProPreO in the Integrated Technology Resource for Biomedical Glycomics.
4.1 Semantically-mediated representation of glycan structures, description of glycan functions
The synthesis of glycans is a complex biochemical process, which is described as a set of metabolic pathways. A complex glycan is synthesized in several steps, each of which should be described in the ontology. The complex metabolic pathways and the single reactions that lead from one glycan to another are modeled to infer similar processes that might lead to the formation of similar glycans that have not yet been discovered or classified.
Figure 7: GlycO representation
of a step in the
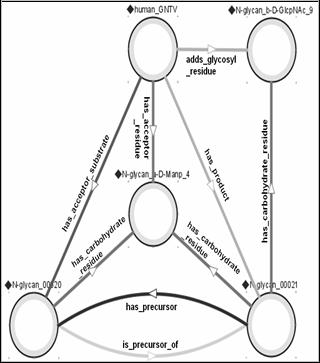 N-Glycan biosynthesis pathway.
N-Glycan biosynthesis pathway.
Glycans are represented as collections of interconnected monosaccharide residues, which are, in turn, classified according to their chemical context within the glycan structure. For example, a typical N-glycan contains a single β-d-Manp residue in its core. This residue is glycosidically linked to a specific site (oxygen-4) of the next residue, which is invariably a β-d-GlcpNAc residue. The identity of the β-d-Manp residue and its precise location in the core of the glycan allows it to be unambiguously classified. In fact, it is often referred to as "the core β-Man residue". The trained glycobiologist intuitively makes a large number of inferences when this colloquial name is invoked, such as correlations between N-glycan branching patterns and biosynthetic mechanisms. However, very few of the residues that make up N-glycans have a common name based on their identity and chemical context.
By modeling the GlycoTree structure, we built a mechanism by which glycans can be semantically classified (as suggested above) simply by checking their constituent (canonical) residues against residue lists, each of which corresponds to a specific type of glycan (e.g., high-mannose or complex N-glycan). Furthermore, the chemical and biological properties of each residue within the glycan, as well as the cellular machinery involved in its biosynthesis and degradation can be semantically inferred. That is, other biological objects (such as glycosyl transferases) and processes (such as metastasis) can be associated with canonical residues that they depend on or interact with. Some of these associations may be indirect (via other objects in the ontology), or inferred by analysis of quantitative information (e.g. correlation of the abundance of glycans containing a specific canonical residue and the observation of a cellular property like invasiveness) that is contained in a semantically annotated database. An example is the specification (within GlycO) that addition of "N-glycan_b-D-GlcNAc_9" is catalyzed by an instance of the GNT-V class of glycosyl transferases (see Figure 7), and that structures elaborated when this residue is present are recognized by the lectin LPHA.
4.2 Semantically-mediated implementation of Glycoproteomics workflow
We are also implementing an ontology-mediated glycoproteomics workflow using ProPreO and GlycO. The aim of this semantic workflow is to enable two tasks from a glycoproteomics research perspective, i.e., ability to correlate and compare two datasets.
We describe a specific example for the use of relationships to correlate relevant datasets. Mass spectrometry phase generates a list of glycopeptides and their relative abundances from a given sample. Real Time Polymerase Chain Reaction (RT-PCR) generates data on the expression of genes for the same sample type. One of the aims of glycoproteomics is to correlate the identification and quantification of glycopeptides with the expression levels of specific genes. This correlation between datasets using an ontological framework is possible by using the extensive set of relationships in GlycO and ProPreO.
To compare data, using the concept of provenance from [40] [41], as discussed earlier, we have developed a semantic data provenance scheme using URIs added at each task in the workflow. Hence, for example, after five tasks in the workflow, the URI of the dataset at the end is constituted of five 'blocks' of URIs. This composite URI for the dataset may be broken down into individual URI blocks that map to a concept in ProPreO. This enables a computational tool to extract information regarding the dataset at any specified stage of the experiment in a single step and use the ontology concept in semantically comparing it to another relevant dataset. This semantic data provenance scheme is flexible in use (single step operation to get provenance information for any specific task in the experiment) and extensible (URI for a dataset is composed of 'blocks' of URIs that are incrementally added at each task in the experiment) in prefix or suffix mode.
We are currently working on annotating the experimental data using concepts from ProPreO and GlycO [28]. The annotation is at two levels, i.e., creation of metadata that is tied to each dataset and annotation of data itself.
5. RELATED WORK
There has been increased activity in development and integration of ontologies. MGED [35] and TAMBIS [34] are built for annotation and integration, respectively. The BioPAX [15] ontology creates a data exchange format for biological pathway data. Most ontologies are built monolithically, but some groups are aiming at building sets of inter-related ontologies. The Open Biomedical Ontologies project [14] and the Gene Ontology Consortium [16] are an example of two related efforts for developing a coherent set of ontologies for this domain.
Current methodological research on building ontologies focuses on the gathering and conceptualization of knowledge while avoiding fallacies in the formal specification of the model [33] [22] [26]. See Jones et al [18] and Cristani/Cuel [4] for extensive surveys on general methodologies such as TOVE, CommonKADS and OTK. A concrete guideline for the development of ontologies is given in Ontology Development 101 by Noy and McGuinness [24]. Schulze-Kremer presents in [29] helpful strategies for discriminating levels in class hierarchies. An excellent guideline to creating semantically sound ontologies is given in the OntoClean methodology [6]. These methodologies help us apply logical rigor to the development, and ease maintenance as well as integration. However, they can not give us insight into how to meaningfully conceptualize the domain of interest.
6. CONCLUSION
Consistency in ontology schema design is essential. Developing two ontologies in the same domain helped us gain an interesting perspective in the design of schemas for different goals. While for the development of GlycO the focus was on building a representation that expressively reflects actual glycan structure and is meant as a basis for reasoning on these structures, the challenge faced in developing ProPreO was that of how to provide a unified interface to distributed heterogeneous data. By adopting OWL-DL for ontology development, we ensured consistency and allowed reasoning tasks to be performed on our ontologies. As stated [32], ontologies will increasingly form the basis of computational tools for solving bioinformatics issues in high-throughput biological experiments. Hence, logical consistency in an ontology is integral to their use by these computational tools.
Relationships play a key role in the usability of biological ontologies. We designed our ontologies with added emphasis on modeling the extensive and rich relationships inherent in the biological domain. This is demonstrated in section 3, as both GlycO and ProPreO feature rich relationships as compared to the MGED ontology.
Population of an ontology connects the schema to real world entities and enables its optimal usage. In our work we also presented the multiple methods used in populating complex ontologies like GlycO and ProPreO. We demonstrated the use of manual and automatic methods for data extraction from heterogeneous and overlapping data sources, in case of GlycO and a dynamic reference resolution to provide a logically unified view of the instance base for extremely large data, in case of ProPreO.
Numerical evaluation of ontologies can only give us structural characteristics. Whether an increase in connectivity and a broader or deeper hierarchy are desirable depends on the user or the task at hand. We were aiming at developing highly connected ontologies and the metrics used show, in this respect, we are at the upper end of current biomedical ontologies.
Building ontologies is still seen as an art or a craft rather than an engineering task. Most likely, the best we can do is to help the knowledge engineers be more efficient in their task by providing tools and a conceptual framework of guidelines for their use. Our work forms a use case that focuses on maintaining consistency, highlights the importance of modeling relationships and population of complex ontologies in the biological domain.
7. ACKNOWLEDGEMENT
This work is part of the Integrated Technology Resource for Biomedical Glycomics (5 P41 RR18502-02), funded by the National Institutes of Health National Center for Research Resources. Donation by Semagix of its Freedom platform for semantic application development is also acknowledged.
Special thanks to Dr. James A. Atwood III and Cory Henson for their contribution and participation in the development of ProPreO and GlycO respectively.
[2] M. Ashburner, CA Ball, J. A. Blake, D Botstein, H. Butler, J. M. Cherry, A. P. Davis, K.Dolinski, S. S. Dwight, J. T. Eppig, M. S. Harris, D.P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J.C. Matese, J. E. Richardson, M. Ringwald, G. M. Rubin, G. Sherlock, Gene Ontology: Tool for the Unification of Biology. Nature Genetics, 25:25-29, 2000.
[3] A. Bohne-Lang, T. Lang, E. Forster, C. W. von der Lieth, 2001. LINUCS: linear notation for unique description of carbohydrate sequences. Carbohydr Res. 336:1-11
[4] M. Cristani and R. Cuel: A Survey on Ontology Creation Methodologies. Int. J. Semantic Web Inf. Syst. 1(2): 49-69 (2005)
[5] S. Doubet and P. Albersheim, CarbBank. Glycobiology, 2, 1992, 505
[6] N. Guarino and C. Welty, "Evaluating Ontological Decisions with OntoClean," Comm. ACM, vol. 45, no. 2, 2002, pp. 61-65.
[7] R. Guha and R. McCool, The tap knowledge base. http://tap.stanford.edu/
[8] I. Horrocks, P. F. Patel-Schneider and F. van Harmelen, From SHIQ and RDF to OWL: the making of a Web Ontology Language, Journal of Web Semantics 1(1): 7-26 (2003)
[9] http://www.genome.ad.jp/kegg/
[10] http://www.glycosciences.de/sweetdb/index.php
[11] http://ncbi.nlm.nih.gov subdirectory /repository/carbbank
[12] http://www.glycosciences.de/tools/linucs/
[13] http://www.daml.org/2003/11/swrl/
[14] http://obo.sourceforge.net/
[16] http://www.geneontology.org/
[17] IUPAC Commission on the Nomenclature of Organic Chemistry (CNOC) and IUPAC-IUB Commission on Biochemical Nomenclature (CBN). Nomenclature of Cyclitols. Recommendations, 1973. Biochem J. 1976 Jan 1;153(1):23-31
[18] D. M. Jones, T. J. M. Bench-Capon, and P.R.S. Visser, Methodologies for Ontology Development. In J. Cuena, editor, Proc. ITi and KNOWS Conference of the 15th IFIP World Computer Congress, pages 62-75, London, UK, 1998. Chapman and Hall Ltd.
[19] M. Kanehisa and S. Goto, KEGG: Kyoto Encyclopedia of Genes and Genomes, Nucleic Acids Research, 2000, Vol. 28, No. 1 27-30
[20] A. Loß, P. Bunsmann, A. Bohne, A. Loß, E. Schwarzer, E. Lang, and C. W. Von der Lieth, SWEET-DB: an attempt to create annotated data collections for carbohydrates, Nucleic Acids Research, 2002 January 1; Vol 30, No. 1, 405-408.
[21] NCRR Integrated Technology Resource for Biomedical Glycomics:http://lsdis.cs.uga.edu/projects/glycomics/, http://cell.ccrc.uga.edu/world/glycomics/researchprogram.php)
[22] I. Niles, A. Pease, Towards a standard upper ontology. In: In Proceedings of the 2nd International Conference on Formal Ontology in Information Systems (FOIS-2001), Chris Welty and Barry Smith, eds. (2001) 17-19.
[23] N. F. Noy, M. Sintek, S. Decker, M. Crubezy, R. W. Fergerson, & M. A. Musen, Creating Semantic Web Contents with Protege-2000. IEEE Intelligent Systems 16(2):60-71, 2001
[25] D. Ramachandran, P. Reagan, K. Goolsbey, First-Orderized ResearchCyc: Expressivity and Efficiency in a Common-Sense Ontology. In Papers from the AAAI Workshop on Contexts and Ontologies: Theory, Practice and Applications. Pittsburgh, Pennsylvania, July 2005.
[26] M. Sabou, C. Wroe, C. Goble and G. Mishne, Learning Domain Ontologies for Web Service Descriptions: an Experiment in Bioinformatics in Proc. 17th Intl Conference on World Wide Web WWW2005, Japan, May 2005
[27] S. S. Sahoo, A. P. Sheth, W. S. York, J. A. Miller, "Semantic Web Services for N-Glycosylation Process", International Symposium on Web Services for Computational Biology and Bioinformatics, VBI, Blacksburg, VA, May 26-27, 2005.
[28] S. S. Sahoo, C. Thomas, A. Sheth, C. Henson, W. S. York, GLYDE-an expressive XML standard for the representation of glycan structure., Carbohydr Res. 2005 Dec 30;340(18):2802-7. Epub 2005 Oct 20. PMID: 16242678
[29] S. Schulze-Kremer, Ontologies for molecular biology and bioinformatics, In Silico Biology 2, 0017 (2002)
[30] A. Sheth, C. Bertram, D. Avant, B. Hammond, K. Kochut, Y. Warke, Managing Semantic Content for the Web, IEEE Internet Computing, July/August 2002, pp. 80-87.
[31] A. Sheth, I. B. Arpinar, and V. Kashyap, Relationships at the Heart of Semantic Web: Modeling, Discovering, and Exploiting Complex Semantic Relationships, in Enhancing the Power of the Internet: Studies in Fuzziness and Soft Computing, M. Nikravesh, L. A. Zadeh, B. Azvine, R. R. Yager (Eds), Springer-Verlag, 63-94
[33] R. Stevens, C. A. Goble and S. Bechhofer, Ontology-based Knowledge Representation for Bioinformatics, Briefings in Bioinformatics. 2000 Nov;1(4):398-414.
[35] C. J. Stoeckert Jr, H. C. Causton & C. A. Ball, Microarray databases: standards and ontologies., Nature Genetics, 32 Supplement - Chipping Forecast II, (December 2002), pp. 469-473
[36] SUMO: http://ontology.teknowledge.com/
[37] N. Takahashi and K. Kato, GlycoTree, Trends in Glycoscience and Glycotechnology, 15, 2003: 235-251.
[38] S. Tartir, I. B. Arpinar, M. Moore, A. Sheth, B. Aleman-Meza, OntoQA: Metric-Based Ontology Quality Analysis, IEEE ICDM 2005 Workshop on Knowledge Acquisition from Distributed, Autonomous, Semantically Heterogeneous Data and Knowledge Sources. Houston, Texas, November 27, 2005
[39] C. F. Taylor et al. "A systematic approach to modeling, capturing, and disseminating proteomics experimental data", Nat. Biotechnol. 2003 Mar; 21(3):247-54
[40] J. Zhao, C. Goble and R. Stevens, Semantic Web Applications to E-Science in Silico Experiments In Thirteenth International World Wide Web Conference (WWW2004) pp. 284-285, New York, May 2004
[41] J. Zhao, C. Wroe, C. Goble, R. Stevens, D. Quan, M. Greenwood, Using Semantic Web Technologies for Representing e-Science Provenance in Proc. 3rd International Semantic Web Conference ISWC2004, Hiroshima, Japan, 9-11 Nov 2004, Springer LNCS 3298