Chemists search for chemical entities (names and formulae) on the Web [19]. We outline how a chemical entity search engine can be built to enable such searches. We conjecture that asimilar approach can be utilized to construct other vertical,domain-specific search engines that need to recognize specialentities.
Users search for chemical formulae using different forms or semantics of the same molecule, e.g., they may search for `CH4' or for `H4C'. Furthermore, users use substrings of chemical names in their search, especially, for well-known functional groups, e.g.,they may search for ``ethyl" to search for the name in Figure4. General-purpose search engines usually support exact keyword searches and do not return documents where only parts of query keywords occur. In this work, we introduce multiple query semantics to allow partial and fuzzy searches for chemical entities. To enable fast searches, we show how to build indexes on chemical entities.
Our investigation shows that end-users performing chemical entity search using a generic search engine, like Google, do not get very accurate results. One of the reasons is that general-purpose search engines do not discriminate between chemical formulae and ambiguous terms. Our search engine identifies chemical formulae and disambiguates its occurrences from those of other strings that could potentially be valid chemical formulae, e.g., ``He", but are actually not. Disambiguating terms is a hard problem because it requires context-sensitive analysis.
A generic search engine does not support document retrieval using substring match query keywords. If we want to enable this kind of searches, a naive approach is to construct an index of all possible sub-terms in documents. Such an index will be prohibitively large and expensive. To support partial chemical name searches, our search engine segments a chemical name into meaningful sub-terms automatically by utilizing the occurrences of sub-terms in chemical names. Such segmentation and indexing allow end-users to perform fuzzy searches for chemical names, including substring search and similarity search. Because the space of possible substrings of all chemical names appearing in text is extremely large, an intelligent selection needs to be made as to which substrings should be indexed. We propose to mine independent frequent patterns, use information about those patterns for chemical name segmentation, and then index sub-terms. Our empirical evaluation shows that this indexing scheme results insubstantial memory savings while producing comparable query results in reasonable time. Similarly, index pruning is also applied to chemical formulae. Finally, user interaction is provided so that scientists can choose the relevant query expansions. Our experiments also show that the chemical entity search engine outperforms general purpose search engines (as expected).
Constructing an accurate domain-specific search engine is a hard problem. For example, tagging chemical entities is difficult due to noise from text recognition errors by OCR, PDF transformation,etc. Chemical lexicons are often incomplete especially with respect to newly discovered molecules. Often they do not contain all synonyms or do not contain all the variations of a chemical formula. Understanding the natural language context of the text to determine that an ambiguous string is chemical entity or not(e.g., ``He" may actually refer to `Helium' as opposed to the pronoun) is also a hard problem to solve with very high precision even if dictionaries of chemical elements and formulae are used.Moreover, many chemical names are long phrases composed of several terms so that general tokenizers for natural languages may tokenize a chemical entity into several tokens. Finally, different representations of a chemical molecule (e.g., ``acetic acid" is written as ``CH3COOH" or ``C2H4O2") have to be taken into account while indexing (or use other strategies like query expansion) to provide accurate and relevant results with a good coverage.
Our search engine has two stages: 1) mining textual chemical molecule information, and 2) indexing and searching for textual chemical molecule information. In the first stage, chemical entities are tagged from text and analyzed for indexing.Machine-learning-based approaches utilizing domain knowledge perform well, because they can mine implicit rules as well as utilize prior knowledge. In the second stage, each chemical entity is indexed. In order to answer user queries with different search semantics, we have to design ranking functions that enable these searches. Chemical entities and documents are ranked using our ranking schemes to enable fast searches in response to queries from users. Synonyms or similar chemical entities are suggested to the user,and if the user clicks on any of these alternatives, the search engine returns the corresponding documents.
Our prior work on chemical formulae in the text[25], while this paper focuses approaches to handle chemical names. We show how a domain-specific chemical entitysearch engine can be built that includes tasks of entity tagging,mining frequent subsequences, text segmentation, index pruning,and supporting different query models. Some previous work is in[25]. We propose several novel methods for chemical name tagging and indexing in this paper. The major contributionsof this paper are as follows:
The rest of this paper is organized as follows: Section 2 reviews related works. Section 3 presents approaches of mining chemical names and formulas based on CRFs and HCRFs. We also propose algorithms for independent frequent subsequence mining and hierarchical text segmentation. Section 4 describes the index schemes, query models, and ranking functions for name and formula searches. Section 5 presents experiments and results. Conclusions and future directions are discussed in Section 6. Web service of our system is provided online1.
Banville has provided a high-level overview on mining chemical structure information from the literature [12]. Hidden Markov Models (HMMs) [9] are one of the common methods for entity tagging. HMMs have strong independence assumptions and suffer from the label-bias problem.Other methods such as Maximum Entropy Markov Models (MEMMs) [16] and Conditional Random Fields (CRFs) [11] have been proposed. CRFs are undirected graph models that can avoid the label-bias problem and relax the independence assumption. CRFs have been used in many applications, such as detecting names[17], proteins [22], genes[18], and chemical formulae [25].Other methods based on lexicons and Bayesian classification have been used to recognize chemical nomenclature [26].The most common method used to search for a chemical molecule is substructure search [27], which retrieves all molecules with the query substructure. However,users require sufficient knowledge to select substructures to characterize the desired molecules for substring search, so similarity search[27,29,23,21]is desired by users to bypass the substructure selection.Other related work involves mining for frequent substructures[8,10] or for frequent sub-patterns[28,30]. Some previous work has addressed how to handle entity-oriented search[6,15]. Chemical markup languages have been proposed [20] but are still not in wide use.None of the previous work focuses on the same problem as ours.They are mainly about biological entity tagging or chemical structure search. We have shown how to extract and search for chemical formulae in text [25], but we propose several new methods to extract and search for chemical names in text documents.
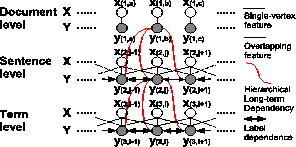
Because we are building a chemical entity search engine,accurately tagging entities in text is important. CRFs[11] is a powerful method for tagging sequential data. We also demonstrate how the accuracy can be improved by considering long-term dependency in the surrounding context at different levels using hierarchical models of CRFs.
Conditional Random Fields
Suppose we have a training set ![]() of
labelled graphs. Each graph in
of
labelled graphs. Each graph in ![]() is an
i.i.d. sample [11]. CRFs
model each graph as an undirected graph
is an
i.i.d. sample [11]. CRFs
model each graph as an undirected graph![]() . Each vertex
. Each vertex ![]() has
a label
has
a label ![]() and an observation
and an observation
![]() . Each edge
. Each edge
![]() represents the
mutual dependence of two labels
represents the
mutual dependence of two labels ![]() . For each sample, the conditional probability
. For each sample, the conditional probability
![]() , where
, where
![]() is the observation
vector of all vertices in
is the observation
vector of all vertices in ![]() ,
,
![]() is the label vector,
and
is the label vector,
and ![]() is the parameter vector
of the model,represents the probability of
is the parameter vector
of the model,represents the probability of ![]() given
given ![]() . An exponential probabilistic model based on
feature functions is used to model the conditional
probability,
. An exponential probabilistic model based on
feature functions is used to model the conditional
probability,
For sequential data, usually chain-structured CRF models are
applied, where only the labels (![]() and
and ![]() ) of
neighbors in a sequence are dependent. Moreover, usually only
binary features are considered. There are two types of
features,state features
) of
neighbors in a sequence are dependent. Moreover, usually only
binary features are considered. There are two types of
features,state features
![]() to
consider only the label (
to
consider only the label (![]() ) of
a single vertex and transition features
) of
a single vertex and transition features
![]() to
consider mutual dependence of vertex labels (
to
consider mutual dependence of vertex labels (![]() and
and![]() ) for each
edge
) for each
edge ![]() in
in ![]() .
State features include two types: single-vertex features obtained
from the observation of a single vertex and overlapping features
obtained from the observations of adjacent vertices. Transition
features are combinations of vertex labels and state features.
Each feature has a weight
.
State features include two types: single-vertex features obtained
from the observation of a single vertex and overlapping features
obtained from the observations of adjacent vertices. Transition
features are combinations of vertex labels and state features.
Each feature has a weight ![]() to specify if the corresponding feature is favored. The weight
to specify if the corresponding feature is favored. The weight
![]() should be highly
positive if feature
should be highly
positive if feature ![]() tends to be
``on" for the training data, and highly negative if it tends to
be ``off".
tends to be
``on" for the training data, and highly negative if it tends to
be ``off".
The log-likelihood for the whole training set ![]() is given by
is given by
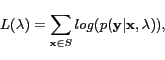 |
(2) |
Hierarchical CRFs
Although CRFs can model multiple and long-term dependencies on the graph and may have a better performance [14] than without considering those dependencies, in practice only short-term dependencies of each vertex (i.e. a word-occurrence)are considered due to the following reasons: 1) usually we do not know what kind of long-term dependencies exist, 2) too many features will be extracted, if all kinds of long-term features are considered, and 3) most long-term features are too sparse and specific to be useful.
However, long-term dependencies at high levels may be useful to improve the accuracy of tagging tasks. For example, at the document level, biological articles have much smaller probabilities of containing chemical names and formulae. At the sentence level, sentences in different sections have different probabilities and feature frequencies of the occurrence of chemical names and formulae, e.g., references seldom contain chemical formulae. Based on these observations, we extend CRFs to introduce Hierarchical Conditional Random Fields (HCRFs) as illustrated in Figure 1. HCRFs start from the highest level to the lowest level of granularity, tag each vertex (e.g.document, sentence, or term) with labels, and use labels as features and generates new features as the interaction with low-level features. At each level, either unsupervised or supervised learning can be used.
The probability models of HCRFs from the highest level to the
level ![]() for a sequence are defined
as
for a sequence are defined
as
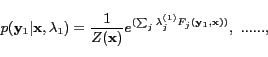 |
(3) |
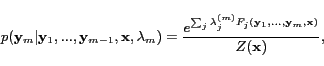 |
(4) |
![]() ,and
,and
![]() ,
,
there are two types of features regarding the higher levels of
label sequences
![]() ,non-interactive
features:
,non-interactive
features:
![]() and
and
![]()
which have no interaction with the observation
sequence![]() , and
interactive features:
, and
interactive features:
![]() ,
,
![]()
that interact with the observation![]() . Interactive features are generated by the
combination of the non-interactive features and the normal
features at the level
. Interactive features are generated by the
combination of the non-interactive features and the normal
features at the level ![]() . For example for
the vertex
. For example for
the vertex ![]() at the level
at the level![]() ,
,
![]() ,where
,where
![]() and
and ![]() are state feature vectors for each vertex
with sizes of
are state feature vectors for each vertex
with sizes of ![]() and
and
![]() ,
and
,
and![]() is a
is a ![]() by
by ![]() matrix of features.
matrix of features.
Feature Set
For entity tagging, our algorithm extracts two categories of
state features from sequences of terms: single-term features, and
overlapping features from adjacent terms. Single-term features
are of two types: surficial features and advanced features.
Surficial features can be observed directly from the term,
e.g.InitialCapital, AllCapitals, HasDigit, HasDash,
HasDot,HasBrackets, HasSuperscripts, IsTermLong.Advanced features
are generated by complex domain knowledge or other data mining
methods, e.g. POS tags learned by natural language parsers, if a
term follows the syntax of chemical formulae, etc. For chemical
name tagging, we use a lexicon of chemical names collected online
by ourselves and WordNet[4]. We support fuzzy
matching on the Levenshtein Distance [13]. Furthermore, we
check if a term has sub-terms learned from the chemical lexicon
based the method in Section 3.5. For sentence tagging, feature
examples are: ContainTermInList,ContainTermPattern,
ContainSequentialTermPattern. For example, a list of journals, a
list of names, and string patterns in reference, are used for
sentence tagging.Overlapping features of adjacent terms are
extracted. We used -1,0, 1 as the window of tokens, so that for
each token, all overlapping features of last token and next token
are included in the feature set. For example, for He in
``.... He is ...", feature(
![]() )=true,
and feature(
)=true,
and feature(
![]() )=true,
where
)=true,
where ![]() means that ``is" is a
``present tense verb, 3rd person singular". The``He" is
likely to be an English word instead ofHelium.
means that ``is" is a
``present tense verb, 3rd person singular". The``He" is
likely to be an English word instead ofHelium.
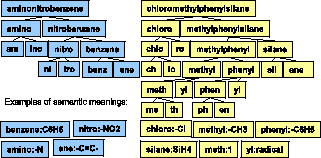
In the last subsection, we use sub-terms as features for chemical name tagging. Moreover, our system supports searches for keywords that occur as parts of terms in documents, e.g., documents with the term "methylethyl" should be returned in response to an user search for ``methyl". Instead of manually discovering those chemical sub-terms by domain experts, we propose unsupervised algorithms to find them automatically from a chemical lexicon and documents.
To support such searches, our system maintains an index of sub-terms. Before it can index sub-terms, the system must segment terms into meaningful sub-terms. Indexing all possible subsequences of all chemical names would make the index too large resulting in slower query processing. Furthermore, users will not query using all possible sub-terms of queries, e.g., a query using keyword ``hyl" is highly unlikely if not completely improbable,while searches for ``methyl" can be expected. So, indexing ``hyl"is a waste. Therefore, ``methyl" should be in the index, while``hyl" should not. However, our problem has additional subtlety.While ``hyl" should not be indexed, another sub-term ``ethyl" that occurs independently of ``methyl" should be indexed. Also, when the user searches for ``ethyl", we should not return the documents that contain ``methyl". Due to this subtlety, simply using the concepts of closed frequent subsequences and maximal frequent subsequences as defined in [28,30] is not enough. We introduce the important concept of independent frequent subsequence. Our system attempts to identify independent frequent subsequences and index them.
In the rest of this paper, we use``sub-term" to refer to a substring in a term that appears frequently. Before our system can index the sub-terms, it must segment chemical names like``methylethyl" automatically into ``methyl" and ``ethyl". The independent frequent sub-terms and their frequencies can be used in hierarchical segmentation of chemical names (explained in details in the next subsection).
First, we introduce some notations as follows:

![]()
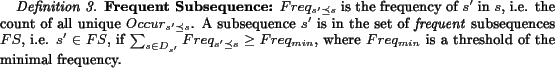
However, usually a sub-term is a frequent subsequence, but not for inverse, since all subsequences of a frequent subsequence are frequent, e.g. ``methyl" (-CH3) is a meaningful sub-term, but``methy" is not, although it is frequent too. Thus, mining frequent subsequences results in much redundant information. We extend two concepts from previous work[28,30] to remove redundant information:
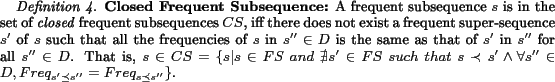
![]()
The example in Figure 3 demonstrates the
set ofClosed Frequent Subsequences, ![]() , and Maximal Frequent Subsequences,
, and Maximal Frequent Subsequences,
![]() . Given the
collectionD={abcde, abcdf, aba, abd, bca} and
. Given the
collectionD={abcde, abcdf, aba, abd, bca} and ![]() =2,support
=2,support ![]() ={abcde,
abcdf},
={abcde,
abcdf},![]() ={abcde, abcdf, aba,
abd}, etc. The set of frequent subsequence FS={abcd, abc,
bcd, ab, bc, cd}has much redundant information. CS={abcd,
ab, bc}removes some redundant information, e.g.
abc/bcd/cd only appears in abcd. MS={abcd}
removes all redundant information as well as useful information,
e.g.,
={abcde, abcdf, aba,
abd}, etc. The set of frequent subsequence FS={abcd, abc,
bcd, ab, bc, cd}has much redundant information. CS={abcd,
ab, bc}removes some redundant information, e.g.
abc/bcd/cd only appears in abcd. MS={abcd}
removes all redundant information as well as useful information,
e.g., ![]() and
and ![]() have occurrences excluding those in
have occurrences excluding those in ![]() . Thus, we need to determine if
. Thus, we need to determine if ![]() and
and ![]() are still
frequent excluding occurrences in
are still
frequent excluding occurrences in ![]() . For a frequent sequence
. For a frequent sequence ![]() ,
its subsequence
,
its subsequence ![]() is also
frequent independently, only if the number of all the occurrences
of
is also
frequent independently, only if the number of all the occurrences
of ![]() not in any occurrences
of
not in any occurrences
of![]() is larger than
is larger than ![]() . If a subsequence
. If a subsequence ![]() has more than one frequent super-sequences, then all the
occurrences of those super-sequences are excluded to count the
independent frequency of
has more than one frequent super-sequences, then all the
occurrences of those super-sequences are excluded to count the
independent frequency of ![]() ,
,
![]() .Thus, in
.Thus, in ![]() ,
, ![]() is frequent and
is frequent and
![]() is frequent independently, but
is frequent independently, but
![]() is not, since
is not, since ![]() occurs twice independently, but
occurs twice independently, but ![]() only once independently. Thus, we get anew set of
independent frequent subsequences
only once independently. Thus, we get anew set of
independent frequent subsequences ![]() .We define independent frequent subsequences
below:
.We define independent frequent subsequences
below:

Why is computing CS or MS not sufficient for our
application?Consider the case of the sub-terms ``methyl" and
``ethyl". Both are independent frequent subsequences in chemical
texts, but not a closed or maximally frequent subsequence. For
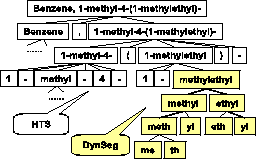
example, for the chemical name in Figure 4,
``methyl" occurs twice and``ethyl" occurs once
independently. Assume in the collection of names
![]() , ``methyl" occurs 100 times,
while ``ethyl" occurs 80times independently. In this case,
``ethyl" is not discovered in MS since it has a frequent
super-sequence ``methyl". In CS,``ethyl" occurs 180 times, since
for each occurrence of ``methyl",a ``ethyl" occurs. Thus, CS
over-estimates the probability of``ethyl". If a bias exists while
estimating the probability of sub-terms, we cannot expect a good
text segmentation result using the probability (next
section).
, ``methyl" occurs 100 times,
while ``ethyl" occurs 80times independently. In this case,
``ethyl" is not discovered in MS since it has a frequent
super-sequence ``methyl". In CS,``ethyl" occurs 180 times, since
for each occurrence of ``methyl",a ``ethyl" occurs. Thus, CS
over-estimates the probability of``ethyl". If a bias exists while
estimating the probability of sub-terms, we cannot expect a good
text segmentation result using the probability (next
section).
Based on these observations, we propose an algorithm for
Independent Frequent Subsequence Mining (IFSM) from a
collection of sequences in Figure 2 with an
example in Figure3. This algorithm considers
sequences from the longest sequence ![]() to the shortest sequence, checking if
to the shortest sequence, checking if ![]() is frequent. If
is frequent. If
![]() , put
, put
![]() in
in ![]() ,
remove all occurrences of its subsequences that are in any
occurrences of
,
remove all occurrences of its subsequences that are in any
occurrences of![]() , and remove all
occurrences overlapping with any occurrences of
, and remove all
occurrences overlapping with any occurrences of ![]() . If the left occurrences of a subsequence
. If the left occurrences of a subsequence
![]() still make
still make![]() frequent, then put
frequent, then put ![]() into
into ![]() . After mining independent
frequent sub-terms, the discovered sub-terms can used as features
in CRFs, and the independent frequencies
. After mining independent
frequent sub-terms, the discovered sub-terms can used as features
in CRFs, and the independent frequencies ![]() can be used to estimate their probabilities for
hierarchical text segmentation in next section.
can be used to estimate their probabilities for
hierarchical text segmentation in next section.
We propose an unsupervised hierarchical text segmentation
method that first uses segmentation symbols to segment chemical
names into terms (HTS in Figure 5), and then
utilizes the independent frequent substrings discovered for
further segmentation into sub-terms (DynSeg in Figure 6). DynSeg finds the best segmentation with the
maximal probability that is the product of probabilities of each
substring under the assumption that all substrings are
independent. After mining the independent frequent substrings to
estimate the independent frequency of each substring ![]() , the algorithm estimates its
probability by
, the algorithm estimates its
probability by
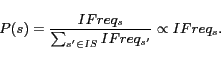 |
(5) |
| (6) |
![\begin{displaymath}P(seg(t))=\prod_{i\in[1,n]}P(s_i),\end{displaymath}](fp50-sun-img84.png) |
(7) |
![\begin{displaymath}L(seg(t))=\sum_{i\in[1,n]}log(P(s_i))).\end{displaymath}](fp50-sun-img85.png) |
(8) |
![\begin{displaymath}seg(t)=argmax_{seg(t)}\sum_{i\in[1,n]}log(P(s_i)).\end{displaymath}](fp50-sun-img86.png) |
(9) |
In practice, instead of segmenting text into ![]() parts directly,usually hierarchical segmentation of
text is utilized and at each level a text string is segmented
into two parts. This is due totwo reasons: 1) It is difficult to
find a proper
parts directly,usually hierarchical segmentation of
text is utilized and at each level a text string is segmented
into two parts. This is due totwo reasons: 1) It is difficult to
find a proper ![]() , and 2) A text string
usually has hierarchical semantic meanings. For example,
``methylethyl" is segmented into ``methyl" and ``ethyl", and then
``methyl" into ``meth" and ``yl", ``ethyl" into ``eth"and ``yl",
where ``meth" means ``one", ``eth" means ``two", and``yl" means
``alkyl".
, and 2) A text string
usually has hierarchical semantic meanings. For example,
``methylethyl" is segmented into ``methyl" and ``ethyl", and then
``methyl" into ``meth" and ``yl", ``ethyl" into ``eth"and ``yl",
where ``meth" means ``one", ``eth" means ``two", and``yl" means
``alkyl".
We discuss two issues in this section: indexing schemes and query models with corresponding ranking functions. For indexing schemes,we focus on how to select tokens for indexing instead of indexing algorithms.
Previous work has shown that small indexes that fit into the main memory have much better query response times [7].We propose two methods of index pruning for the chemical name and formula indexing respectively. For chemical formula indexing, we proposed the strategy in [25].
For chemical name indexing, a naive way of index construction for substring search is to index each character and its position, and during substring search, like phrase search in information retrieval, chemical names containing all the characters in the query string are retrieved and verified whether the query substring appears in the returned names. Even though the index size is quite small using this approach, it is not used due to two reasons: 1) Each character has too many matched results to return for verification, which increases the response time of queries; 2)a matched substring may not be meaningful after text segmentation,e.g. ``methyl" (-CH3) should not be returned when searching for chemical names containing ``ethyl" (-C2H5).These indexed tokens can support similarity searches and most meaningful substring searches as shown in the experiment results in Section 5.4. As mentioned in Subsection 3.3, typically, users use meaningful substring searches.Thus, for a chemical name string, ``methylethyl", indexing``methyl" and ``ethyl" is enough, while ``hyleth" is not necessary. Hence, after hierarchical text segmentation, the algorithm needs to index substrings at each node on the segmentation tree. This reduces the index size tremendously (in comparison with indexing all possible substrings), and our index scheme allows the retrieval of most of the meaningful substrings.If only strings at the high levels are indexed, then nothing is returned when searching for string at lower levels. If only strings at the lowest levels are indexed, too many candidates are returned for verification. Thus, at least, we need to index strings at several appropriate levels. Since the number of all the strings on the segmentation tree is no more than twice of the number of leaves, indexing all the strings is a reasonable approach.
Features based on selected subsequences (substrings in names and partial formulae in formulae) should be used as tokens for search and ranking. Extending our previous work [25], we propose three basic types of queries for chemical name search:exact name search, substring name search, andsimilarity name search. Usually only exact name search and substring name search are supported by current chemistry databases [2]. We have designed and implemented novel ranking schemes by adapting term frequency and inverse document frequency, i.e. TF.IDF, to rank retrieved chemical entities, i.e., names and formulae.
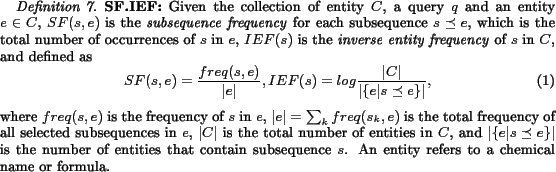
Exact name search
An exact name search query returns chemical names with documents where the exact keyword appears.
Substring name search
Substring name searches return chemical names that
contain the user-provided keyword as a substring with documents
containing those names. If the query string is indexed, the
results are retrieved directly. Otherwise, the query string is
segmented hierarchically, then substrings at each node are used
to retrieve names in the collection, and the intersection of
returned results are verified to check if the query are
contained. This happens rarely since most frequent substrings are
indexed.The ranking function of substring name search regarding a
query![]() and a name string
and a name string ![]() is given as
is given as
| (10) |
Similarity name search
Similarity name searches return names that are similar
to the query. However, the edit distance for similarity
measurement is not used for two reasons: 1) Computing edit
distances of the query and all the names in the data set is
computationally expensive, so a method based on indexed features
of substrings is much faster and feasible in practice. 2)
Chemical names with similar structures may have a large edit
distance. Our approach is feature-based similarity search, where
substring features are used to measure the similarity. We design
a ranking function based on indexed substrings, so that the query
is processed and the ranking score is computed efficiently.
Similar to substring search, first a query string is segmented
hierarchically, then substrings at each node are used to retrieve
names in the collection, and scores are computed and summed up.
Longer substrings are given more weight for scoring, and scores
of names are normalized by their total frequency of substrings.
The ranking function is given as
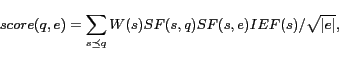 |
(11) |
The chemical formula searches are similar and presented in our prior work [25].Conjunctive searches of the basic name/formula searches are supported, so that users can define various constraints to search for desired names or formulae.When a user inputs a query that contains chemical name and formula searches as well as other keywords, the whole process involves two stages: 1) name/formula searches are executed to find desired names and formulae, and 2) returned names and formulae as well as other keywords are used to retrieve related documents. We useTF.IDF as the ranking function in the second stage, and the ranking scores of each returned chemical name/formula in the first stage are used as term weights to multiple theTF.IDF of each term when computing the ranking score in the second stage.
In this section, our proposed methods are examined. We present and discuss corresponding experimental results.
 |
Since our chemical name tagging uses data mining with features
of domain knowledge. Some features are based on a chemical
lexicon.To construct a lexicon of chemical names, we collected
221,145chemical names from multiple chemical webpages and
onlinedatabases.We evaluate our algorithm of IFSM using this
lexicon with different threshold values
![]() .
We first tokenize chemical names and get 66769 unique terms. Then
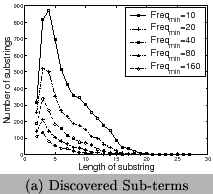
we discover frequent sub-terms from them. The distribution of
sub-term's lengths with different values of
.
We first tokenize chemical names and get 66769 unique terms. Then
we discover frequent sub-terms from them. The distribution of
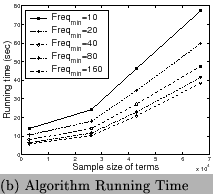
sub-term's lengths with different values of ![]() are presented in Figure 7 (a) and the run time of our algorithm is shown in
Figure 7 (b). Most discovered sub-terms have
semantic meanings in Chemistry. We show some of the frequent
sub-terms with their meanings in Table 1.
After IFSM, hierarchical text segmentation is tested using the
same lexicon and tagged chemical names from online documents, and
examples of the results are shown in Figure 8.
are presented in Figure 7 (a) and the run time of our algorithm is shown in
Figure 7 (b). Most discovered sub-terms have
semantic meanings in Chemistry. We show some of the frequent
sub-terms with their meanings in Table 1.
After IFSM, hierarchical text segmentation is tested using the
same lexicon and tagged chemical names from online documents, and
examples of the results are shown in Figure 8.
The data to test tagging chemical entities is randomly selected from publications crawled from the Royal Society of Chemistry[3].200 documents are selected randomly from the data set, and a part of each document is selected randomly to construct the training set manually. The training set is very imbalanced because of the preponderance of non-chemical terms. We first apply CRF and10-fold cross-validation for sentence tagging. We manually construct the training set by labeling each sentence ascontent (content of the documents) or meta(document meta data, including titles, authors, references, etc.).Results for sentence tagging are presented in Table 2.Then sentence tags can be used as features for chemical entity tagging at the term level.
For chemical formula tagging, we use 10-fold cross-validation
and a 2-level HCRF, where the two levels are the sentence level
and the term level. The sentence tags are used as features for
chemical formula tagging. At the term level, we label each token
as a formula or a non-formula. Several methods are
evaluated, including rule-based String Pattern Match, CRFs with
different feature sets, and HCRFs with all features.Features are
categorized into three subsets: features using rule-based string
pattern match (RULE), features using part-of-speech tags
(POS), and other features. Four combinations are tested:
(1) all features, (2) no POS, (3) no RULE, and (4) no POS or
RULE. Results of chemical formula tagging are shown in Table
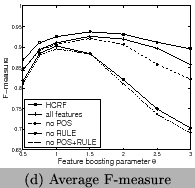
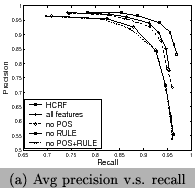
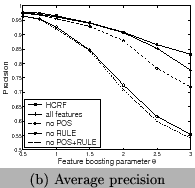
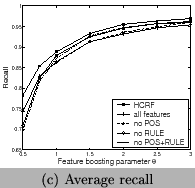
3 and Figure 9. We can
see that the RULE features contribute more than the POS features
and HCRF has the highest contribution. In comparison to CRFs,
HCRFs only has an improvement of ![]() . However, since the total error rate is just
. However, since the total error rate is just
![]() , the improvement is about
, the improvement is about
![]() of the total error rate of
of the total error rate of
![]() . This means
long-dependence features at the sentence level has positive
contributions. HCRF has a better performance than CRF, but
increases the runtime. Both for HCRF and CRF using all features,
the best F-measureis reached when
. This means
long-dependence features at the sentence level has positive
contributions. HCRF has a better performance than CRF, but
increases the runtime. Both for HCRF and CRF using all features,
the best F-measureis reached when ![]() , and in this case recall and precision are
balanced. We show the results using all features in Table
3.
, and in this case recall and precision are
balanced. We show the results using all features in Table
3.
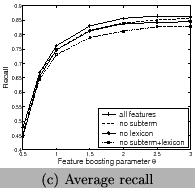
For chemical name tagging, since a name may be a phrase of
several terms, we label each token as a B-name (beginning
of a name), or I-name (continuing of a name), or
anon-name. We use 5-fold cross-validation and CRF with
different feature sets. Features are classified into three
subsets: features using frequent sub-terms
(sub-term),features using lexicons of chemical names and
WordNet[4]
(lexicon), and other features.Four combinations are
tested: (1) all features, (2) no sub-term,(3) no lexicon, and (4)
no sub-term or lexicon. We test different values {0.5, 0.75, 1.0,
1.5, 2.0, 2.5, 3.0} for the feature boosting parameter
![]() for the formula (or
B-name) class.Note that when
for the formula (or
B-name) class.Note that when ![]() , it is the normal CRF, while when
, it is the normal CRF, while when![]() , the non-formula
(or non-name) class gets more preference. To measure the overall
performance, we useF
, the non-formula
(or non-name) class gets more preference. To measure the overall
performance, we useF![]() [18],
where
[18],
where ![]() is precision and
is precision and ![]() is recall, instead of using
error rate, since it is too small for imbalanced data. Results of
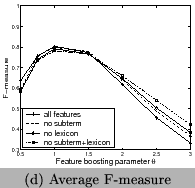
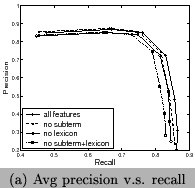
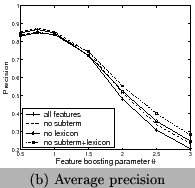
chemical name tagging are shown in Table 4
and Figure 10. We can see using all features
has the best recall and F-measure, and using features of frequent
sub-terms can increase recall and F-measure but decrease
precision. Our system clearly outperforms the tool for chemical
entity tagging: Oscar3 [5] on the same data sets. Oscar3
has a recall of 70.1%,precision of 51.4%, and the
F-measure is59.3%.
is recall, instead of using
error rate, since it is too small for imbalanced data. Results of
chemical name tagging are shown in Table 4
and Figure 10. We can see using all features
has the best recall and F-measure, and using features of frequent
sub-terms can increase recall and F-measure but decrease
precision. Our system clearly outperforms the tool for chemical
entity tagging: Oscar3 [5] on the same data sets. Oscar3
has a recall of 70.1%,precision of 51.4%, and the
F-measure is59.3%.
We only evaluate HCRFs for chemical formula tagging, because we found that ambiguous terms, like ``C", ``He", ``NIH", appearing frequently in parts containing document meta data, have different probabilities to be chemical formulae. For example,``C" is usually a name abbreviation in the part of authors and the references, but usually refers to ``Carbon" in the body of document. Thus, HCRFs is evaluated for chemical formula tagging to check whether the long dependence information at the sentence level is useful or not. HCRFs are not applied for chemical name tagging, because we found that the major source of errors in chemical name tagging is not due to term ambiguity, but due to limitations of the chemical dictionary and incorrect text tokenizing.
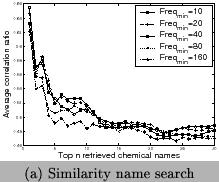
For chemical name indexing and search, we use the segmentation
based index construction and pruning. We compare our approach
with the method using all possible substrings for indexing. We
use the same collection of chemical names in Section 5.1. We
split the collection into two subsets. One is for index
construction (37,656chemical names), while the query names are
randomly selected from the other subset.Different values of the
frequency threshold
![]() to mine independent frequent substrings are tested. The
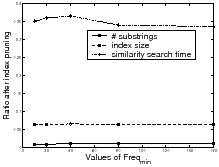
experiment results in Figure11 show that
most (
to mine independent frequent substrings are tested. The
experiment results in Figure11 show that
most (![]() ) substrings are removed
after hierarchical text segmentation, so that the index size
decreases correspondingly (
) substrings are removed
after hierarchical text segmentation, so that the index size
decreases correspondingly (![]() of
the original size left).
of
the original size left).
After index construction, for similarity name search, we
generate a list of 100 queries using chemical names selected
randomly: half from the set of indexed chemical names and half
from unindexed chemical names. These formulae are used to perform
similarity searches. Moreover, for substring name search, we
generate a list of 100 queries using meaningful and the most
frequent sub-terms with the length 3-10 discovered in Section
5.1. We also evaluated the response time for similarity name
search, illustrated in Figure 11. The method
using HTS only requires ![]() of
the time for similarity name search compared with the
method using all substrings.However, we did not test the case
where the index using all substrings requires more space than the
main memory. In that case, the response time will be even
longer.
of
the time for similarity name search compared with the
method using all substrings.However, we did not test the case
where the index using all substrings requires more space than the
main memory. In that case, the response time will be even
longer.
We also show that for the same query of similarity name search
or substring name search, the search result using
segmentation-based index pruning has a strong correlation with
the result before index pruning. To compare the correlation
between them, we use the average of the percentage of overlapping
results for the top![]() retrieved formulae that is defined as
retrieved formulae that is defined as
![]() ,where
,where
![]() and
and ![]() are the search results of before and after index
pruning, respectively. Results are presented in Figure12. We can observe that for similarity search, when
more results are retrieved, the correlation curves decrease,
while for substring search, the correlation curves increase.
When
are the search results of before and after index
pruning, respectively. Results are presented in Figure12. We can observe that for similarity search, when
more results are retrieved, the correlation curves decrease,
while for substring search, the correlation curves increase.
When![]() is larger, the
correlation curves decrease especially for substring search.
is larger, the
correlation curves decrease especially for substring search.
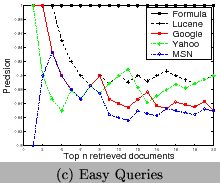
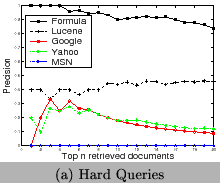
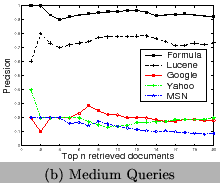
To test the ability of our approach for term disambiguation in documents, we index 5325 PDF documents crawled online. We then design 15 queries of chemical formulae. We categorize them into three levels based on their ambiguity, 1) hard (He, As, I,Fe, Cu), 2) medium (CH4, H2O, O2, OH, NH4), and 3) easy(Fe2O3, CH3COOH, NaOH, CO2, SO2). We compare our approach with the traditional approach by analyzing the precision of the returned top-20 documents. The precision is defined as the percentage of returned documents really containing the query formula. Lucene [1] is used for the traditional approach. We also evaluate generic search engines, Google, Yahoo, and MSN using those queries to show the ambiguity of terms. Since the indexed data are different, they are not comparable with the results of our approach and Lucene. Their results illustrate that ambiguity exists and the domain-specific search engines are desired. From Figure 13, we can observe 1) the ambiguity of terms is very serious for short chemical formulae, 2) results of Google and Yahoo are more diversified than that of MSN, so that chemical web pages are included, and 3) our approach out-performs the traditional approach based on Lucene, especially for short formulae.
We evaluated CRFs for chemical entity extraction and proposed an extended model HCRFs. Experiments show that CRFs perform well and HCRFs perform better. Experiments illustrate that most of the discovered sub-terms in chemical names using our algorithm of IFSM have semantic meanings. Examples show our HTS method works well.Experiments also show that our schemes of index construction and pruning can reduce indexed tokens as well as the index size significantly. Moreover, the response time of similarity name search is considerably reduced. Retrieved ranked results of similarity and substring name search before and after segmentation-based index pruning are highly correlated. We also introduced several query models for name searches with corresponding ranking functions. Experiments show that the heuristics of the new ranking functions work well. In the future,a detailed user study is required to evaluate the results. Entity fuzzy matching and query expansion among synonyms will also be considered.
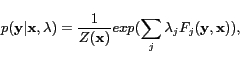
![\begin{figure} \centering \par \framebox[8.4cm][t]{ \parbox{8.1cm}{ \par \smal... ...Occur_{t\preceq s'} \neq \emptyset \wedge t\in IS$. \par } } \par \end{figure}](fp50-sun-img72.png)
![\begin{figure} \centering \par \framebox[8.4cm][t]{ \parbox{8.1cm}{ \par \smal... ...\{abcd, ab\}$, $IFreq_{abcd}=2\ \&\ IFreq_{ab}=2$. \par } } \par \end{figure}](fp50-sun-img74.png)
![\begin{figure} \centering \par \framebox[8.4cm][t]{ \parbox{8.1cm}{ \par \smal... ...Seg}($s$,$IF$,$r$,2); \par 8. \textbf{else return}; \par } } \par \end{figure}](fp50-sun-img88.png)
![\begin{figure} \centering \par \framebox[8.4cm][t]{ \parbox{8.15cm}{ \par \sma... ...space{0.5cm}\textbf{DynSeg}($s'$,$IF$,$r'$,$n$); \par } } \par \par \end{figure}](fp50-sun-img89.png)